Image

Title: Molecular Dynamic Simulations on Mechanism Design for Protein Degradation
IMPACT
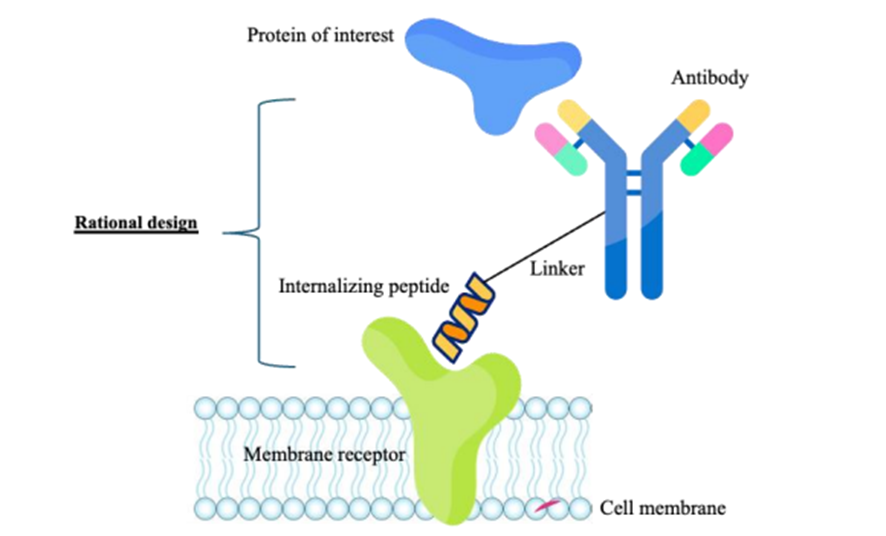
This collaboration had a significant impact by enabling the first computational exploration of LYTAC (Lysosome-Targeting Chimeras) design at a level of detail that would not have been feasible with local resources alone. Access to high-performance computing allowed the team to model complex peptide–glycan–protein systems, providing essential structural and dynamic insights for early-stage targeted protein degradation strategies. The work strengthens Europe’s computational drug-discovery capacity and accelerates the translation of innovative therapeutic concepts into practical applications.
BENEFITS
Enabled the first molecular dynamics (MD) simulations of antibody/peptide–protein complexes relevant to LYTAC candidates.
Allowed assessment of system stability and key interaction patterns.
Provided preliminary exploration of conformational dynamics for early candidate designs.
Established validated protocols and workflows for future drug-design simulations.
These benefits form a crucial computational foundation for advancing LYTAC-based therapeutics toward experimental development.
KEY POINTS BEFORE AGREEING ON THE PROJECT
Before initiating the collaboration, several aspects needed alignment:
- The computational scale required: simulations at atomistic resolution of large receptor–ligand systems exceeded local infrastructure capacity.
- The necessity of HPC expertise for system setup, parameterization, model preparation, and simulation workflow design.
- The need for secure and efficient project coordination between CESGA and LincBiotech.
- Clear definition of project goals: establishing protocols, performing small-scale tests, and validating simulation feasibility rather than delivering full production runs.
- Agreement on resource allocation, timelines, and expected intermediate outcomes.
TECHNICAL/SCIENTIFIC CHALLENGE
The project’s central challenge was to investigate the molecular mechanisms underlying LYTAC design — an emerging technology for targeted protein degradation — by simulating complex biomolecular assemblies at atomic resolution.
These simulations involve peptide–linker–antibody conjugates interacting with receptors and glycans, resulting in extremely large systems with high computational demands. Local computational resources were insufficient to handle the size, stability requirements, and time scales of these simulations, making progress impossible without dedicated HPC support.
SOLUTION
Thanks to the EuroCC2 program and access to CESGA’s HPC infrastructure, the research team was able to:
- Prepare and assemble large LYTAC-related systems.
- Conduct initial molecular dynamics simulations on high-potential building blocks.
- Validate system preparation protocols and refine simulation parameters.
- Perform preliminary stability and equilibration tests for peptide–linker–antibody constructs bound to their receptors.
- Although production-scale simulations have not yet been performed, the collaboration provided the essential groundwork required to reach that stage.
WHAT YOU ACHIEVED TOGETHER THAT COULD NOT BE DONE SEPARATELY
Independently, LincBiotech could not initiate atomistic MD simulations of LYTAC systems due to hardware limitations.
Through the collaboration with CESGA and EuroCC2:
- Large, realistic biomolecular systems could be assembled and equilibrated.
- Early-stage simulations were successfully tested for feasibility.
- The team validated protocols for future large-scale MD campaigns.
This joint effort enabled a level of computational work that would have been entirely unattainable with in-house resources, effectively advancing the drug-design pipeline.